
As
previously discussed on RFN, the single pool Kt/V (spKt/V) was developed by
Gotch and Sargent in a reanalysis of the
NCDS data in an attempt to distinguish the dose of hemodialysis associated with improved outcomes.
The spKt/V model assumes that the human body acts like a “single pool” with a single concentration of urea. The model describes what happens when the dialysis machine is attached to the single pool and removes some of that urea (figure 1).
Each time the fluid comes into the dialyzer, some of the urea is removed and the remainder is returned to the pool. So over time the dialysis machine is fed fluid with progressively lower urea concentrations. As the dialysis treatment proceeds less urea is removed for any given volume.
For example, if at the start of the run the machine is clearing 100 ml/min (1 dl/min) and the concentration of urea is 100 mg/dl, 100 mg of urea will be removed from the body in one minute. However, if towards the end of the run the concentration of urea is 40 mg/dl, only 40 mg of urea will be removed in one minute.

This constant fractional removal leads to a curvilinear decline in the urea concentration during the dialysis run as shown in figure 2 by the solid line. When the urea concentration is expressed as a logarithm the decline becomes linear (dotted line) with a slope of –K/V.
Using the starting BUN, the constant fractional decline and a set amount of time, one can figure out what the final BUN will be…

Remember that K = clearance in ml/min, t = time in minutes, and V = the volume of distribution of urea in liters leading to the dimensionless ratio Kt/V.
Rearranging…
Kt/V = -ln(R)
R = (post dialysis BUN/pre dialysis BUN). Plug in the numbers and see that a post dialysis BUN/pre dialysis BUN of 0.37 gives you a Kt/V of 1.0.
A spKt/V of 1.0 means that the entire of volume of the single pool has passed through the dialyzer once.
The fancy looking Daugardis equation,
mentioned last time for spKt/V, contains additional adjustments for urea generation during the dialysis run and urea removal that occurs from convection (i.e. clearance from ultrafiltration).


The spKt/V predicts urea concentration change as shown by the dashed line in the figure 3. However, what actually happens on dialysis is shown by the dots. The BUN drops faster than predicted during dialysis and rebounds more quickly afterwards.
The net impact is that spKt/V overestimates the amount of urea removed during a dialysis session.

This occurs because the human body is not a single pool of urea. Instead, it has multiple compartments, each with unique rates of urea clearance. During dialysis the intravascular and interstitial spaces are cleared of urea rapidly while the larger intracellular space has lower urea clearance, as diagrammed in figure 4. This leads to the rapid decline in BUN during therapy.

Additionally, the entire blood volume can not be treated as a single compartment. Blood in the cardiopulmonary circuit cycles through the dialysis machine every 10-15 seconds while blood flowing through the peripheral limbs may take several minutes.
After dialysis there is a rebound in urea concentration as intracellular urea leaks into the interstitial and plasma space and blood from poorly perfused regions mixes with the highly dialyzed central circulation.
The equilibrated Kt/V can be obtained by measuring the BUN 30-60 minutes after the end of dialysis. It provides a more accurate measure of dose by using a urea concentration that is more reflective of the concentration in the total body water as compared with spKt/V, which uses a concentration reflective of the extracellular compartment and central circulation.
Of course, having someone stay for an additional hour after their dialysis session ends is inconvenient. Luckily there are conversion equations from spKt/V to eKt/V as shown below (there are several available equations for doing this).
eKt/V = spKt/V [(t/(t + 35)]

Notice the importance of time for any given spKt/V. For example, with spKt/V held constant and varying the time from say 100 minutes to 200 minutes the eKt/V will go from 74% to 85% of the spKtV. The longer you run the closer eKt/V will be to spKt/V (figure 6).
In the Hemodialysis (
HEMO) Study published in 2002, 1846 patients were randomized to either high or low flux dialysis membranes and standard or high dose 3x per week dialysis. The dose targets were in eKt/V calculated from spKt/V. As mentioned above, eKt/V was used because it is a more accurate reflection of dose.
The achieved mean eKt/Vs in the standard dose and high dose groups were 1.16 and 1.53 respectively with mean spKt/Vs in the same groups of 1.32 and 1.71. There were no differences between groups in the primary outcome of death from any cause.
As I’m sure you’ve noted NCDS and HEMO where both trials of 3x week hemodialysis schedules. What if we want to compare dose between more or less frequent dialysis? Up next, stdKt/V…








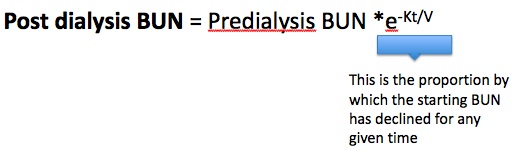







 I was recently called to evaluate an obese patient with profound metabolic acidosis who was admitted to the hospital with change in mental status and poor oral intake for several days. Her admission labs revealed severe hyperglycemia (glucose 776 mg/dL) and normal anion gap hyperchloremic metabolic acidosis (serum pH of 7.11 and serum bicarbonate of 7 mmol/L). She was initially suspected as having diabetic ketoacidosis due to miscalculation of the anion gap by the admitting team who used the corrected serum sodium and not the actual sodium, and as expected, her acidosis did not resolve despite the correction of hyperglycemia with intravenous fluids and insulin.
I was recently called to evaluate an obese patient with profound metabolic acidosis who was admitted to the hospital with change in mental status and poor oral intake for several days. Her admission labs revealed severe hyperglycemia (glucose 776 mg/dL) and normal anion gap hyperchloremic metabolic acidosis (serum pH of 7.11 and serum bicarbonate of 7 mmol/L). She was initially suspected as having diabetic ketoacidosis due to miscalculation of the anion gap by the admitting team who used the corrected serum sodium and not the actual sodium, and as expected, her acidosis did not resolve despite the correction of hyperglycemia with intravenous fluids and insulin. 







