
A 70 year old Ghanaian man was recently admitted under our
care. He had been diagnosed with aggressive myelodysplasia
2 months previously after presenting with fatigue and abnormal blood results (WBC
50.3, platelets 130 and LDH 928 at the time of
diagnosis). A plan was made for
palliative chemotherapy. One month after his diagnosis he developed a large
pericardial effusion and had 1L of haemorrhagic fluid drained. At this point his creatinine was 200 umol/L
(2.26 mg/dL). Routine and TB culture of
the fluid was negative, as was cytology and immunophenotyping.
Two weeks after this admission he represented with abdominal
pain. A CT showed bilateral renal and
bladder calculi without obstruction. He
was oliguric with a creatinine of 577 umol/L (6.5 mg/dL) rising to 709 umol/L (8 mg/dL)
over the next 12 hours. His uric acid
level was 18.0 mg/dL, which had not been checked previously. Phosphate was 1.86 mmol/L (5.75 mg/dL), Ca
2.1 mmol/L (8.4 mg/dL) and K 4.6 mmol/L.
Our diagnosis was of a spontaneous tumour lysis syndrome
(TLS; see previous RFN posts here & here).
Nucleic acids released from tumour cell lysis are broken down into
xanthine and then uric acid by xanthine oxidase. Renal failure is caused by uric acid
precipitating in renal tubules causing a mechanical obstruction and
inflammatory reaction. While TLS is
typically seen following initiation of chemotherapy causing a rapid breakdown
of cancer cells, a spontaneous form has been described in acute leukaemia and
NHL. Our patient was at high risk of
converting into AML but had no rise in peripheral blasts to suggest this.
Interestingly, spontaneous tumour lysis syndrome is
associated with hyperuricemia but often without the hyperphosphatemia (and
hyperkalemia) seen in the classical form of the disease– thought to be because
the released phosphorus is quickly used up in the generation of new tumour
cells. This would fit with our patients
results.
Our patient was commenced on dialysis which gave reductions
in uric acid levels of 50% per treatment, but they quickly rebounded. He was no longer fit for treatment of his
myelodysplasia making longer term management more difficult. Given his African ethnicity, we checked his
glucose-6-phosphatase levels, which were normal, before he received rasburicase
(recombinant urate oxidase). Rasburicase reduces uric acid levels by converting
it into allantoin. It may cause severe
oxidative hemolysis if glucose-6-phosphatase deficient. Uric acid fell to undetectable
levels following this however he had an ongoing dialysis requirement (note that
rasburicase retains in vitro activity in the blood bottle so sample should
ideally go on ice). Allopurinol as a
longer term medication to reduce uric acid formation may be useful, but may not
manage to suppress formation sufficiently.
In addition to tumour lysis syndrome, acute urate
nephropathy can be caused by other states of tissue catabolism such as seizures, in primary overproduction of uric acid or in
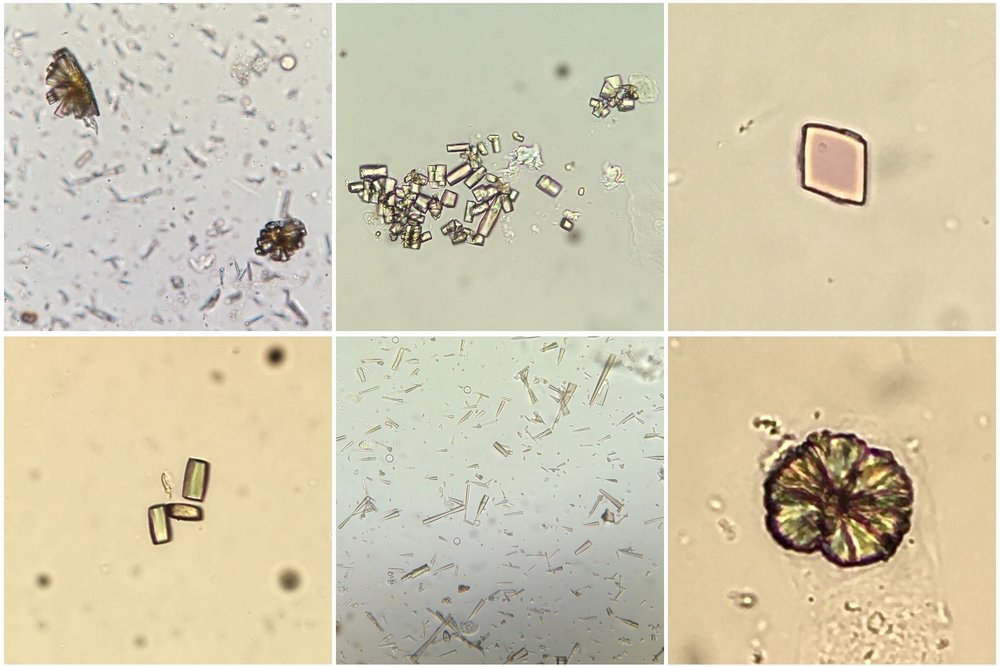
cases of reduced urate reabsorption in the proximal tubule. Urinalysis can show uric acid crystals
(birefringent with polarisation; see image) or can be normal (as in our patient) perhaps due
to a lack of output from obstructed tubules.
This case raised several points to me. Was his pericardial
effusion also caused by a urate infiltration?
No clear cause was ever identified at the time and he did not appear
‘uremic’ despite his renal dysfunction.
Could any of this have been prevented if treatment for his hyperuricemia
had been commenced earlier? I also
learned:
- The nuances of spontaneous tumor lysis syndrome (often phosphate & K not hugely elevated).
- Rasburicase is contraindicated if glucose-6-phosphatase deficient (approximately 20% of Africans).
- The ‘undetectable’ result of urate after rasburicase administration appears to be due to in vitro activity of the drug in the blood bottle.
Image thanks to Florian Buchkremer @swissnephro
Post by Ailish Nimmo














No comments:
Post a Comment