

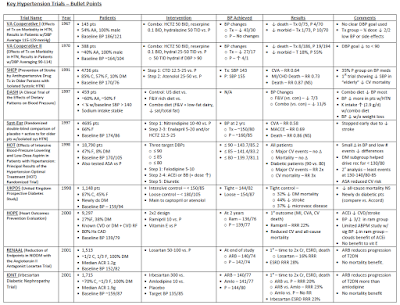
While no trial has directly compared low-dose HCTZ monotherapy with a thiazide-like diuretic, the MRFIT trial offers the closest comparator with the results offering cautionary lessons regarding the benefits of HCTZ at historically prescribed doses (50-100 mg/day). Published in 1982, patients were randomized to placebo or combined lifestyle/drug therapy with HCTZ or chlorthalidone as the initial agent. Not only did HCTZ treated patients have higher all-cause mortality rates compared to those in the chlorthalidone arm but there was a trend towards increased all-cause mortality when HCTZ patients were compared to placebo. Moreover, when those randomized to HCTZ were switched to chlorthalidone, rates of death from coronary artery disease fell below those recorded in the placebo arm.
In contrast, nearly every seminal trial showing improved CV outcomes with diuretics has utilized the thiazide-like diuretics chlorthalidone or indapamide at contemporary daily doses of 25 and 2.5mg, respectively (ADVANCE, HDFP). The SHEP trial was the first large study to address the treatment of isolated systolic hypertension with more aggressive blood pressure goals; compared to placebo, those randomized to chlorthalidone had far fewer CV events. The ALLHAT study, which compared the efficacy of full dose chlorthalidone (25mg/d) with lisinopril (40mg/d), norvasc (10mg/d), and doxazosin (arm terminated prematurely), found that chlorthalidone resulted in the lowest blood pressure among treatment groups. It was not only as effective as the other agents in preventing the primary CV outcome but was superior with respect to the secondary outcome of heart failure (although this may have been related to improved blood pressure control rather than the agent itself). Moreover, truly elderly patients appear to tolerate the more potent thiazide-like diuretics. For example, the HYVET trial was the first study to evaluate the effects of tighter BP control among (relatively healthy) individuals over 80. Those treated with an indapamide-based regimen not only had far fewer fatal strokes but lower rates of adverse events compared to those on placebo.
Small “proof of concept” studies offer possible explanations for these differences in outcome. In a 24-hour ambulatory blood pressure monitoring study comparing equi-potent doses of HCTZ (12.5mg day) and chlorthalidone (6.25mg/day), chlorthalidone distinguished itself by its extended duration of action resulting in blood pressure control throughout the 24-hour period. The pharmacologic property is critical as agents with 24-hour efficacy protect against the morning “surge” in blood pressure, a time when patients are most vulnerable to stroke and myocardial infarction 26821625).
In light of the unequivocal therapeutic efficacy of thiazide-like diuretics and the lack of evidence supporting HCTZ as mono-therapy, the nephrology community should serve as the exception to the aforementioned trend in thiazide prescription patterns. Concern that chlorthalidone/indapamide may be too potent for the elderly are well founded but as the above trials in the aged demonstrate, low-dose therapy (e.g. indapamide 1.25mg/d) can be prescribed, allowing for less volume depletion and metabolic sequelae while providing the extended anti-hypertensive response that may be the key to their superiority over HCTZ.
Conflicts of Interest: None
Hillel Sternlicht, MD
Hypertension Specialist
Author, Concepts in Hypertension Newsletter- Subscribe for free






















{kind=link}